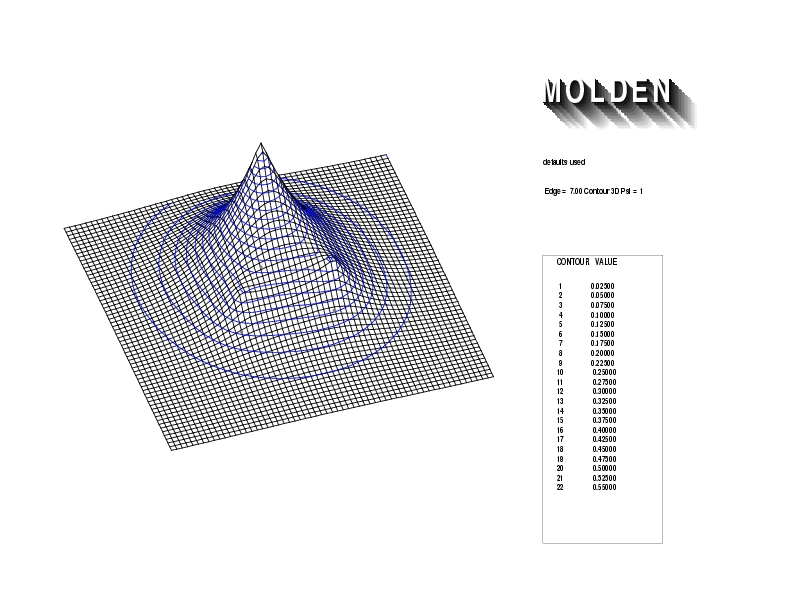
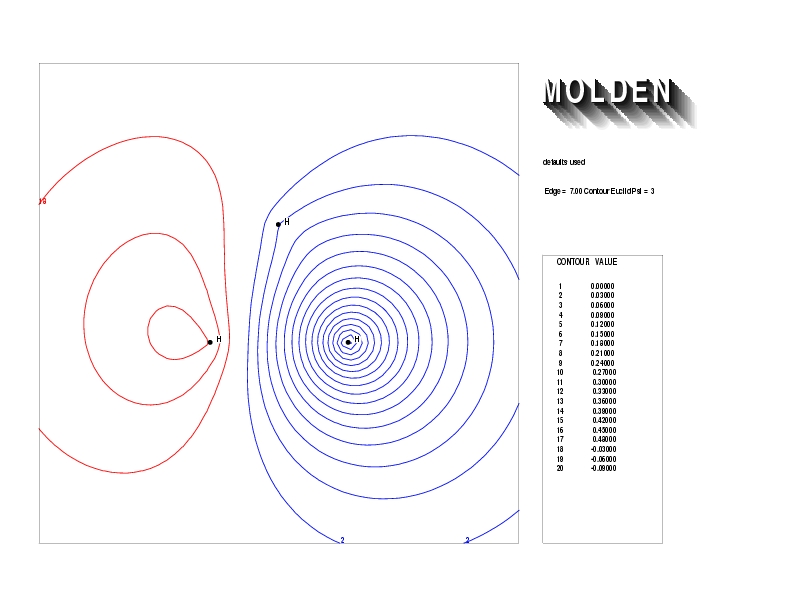
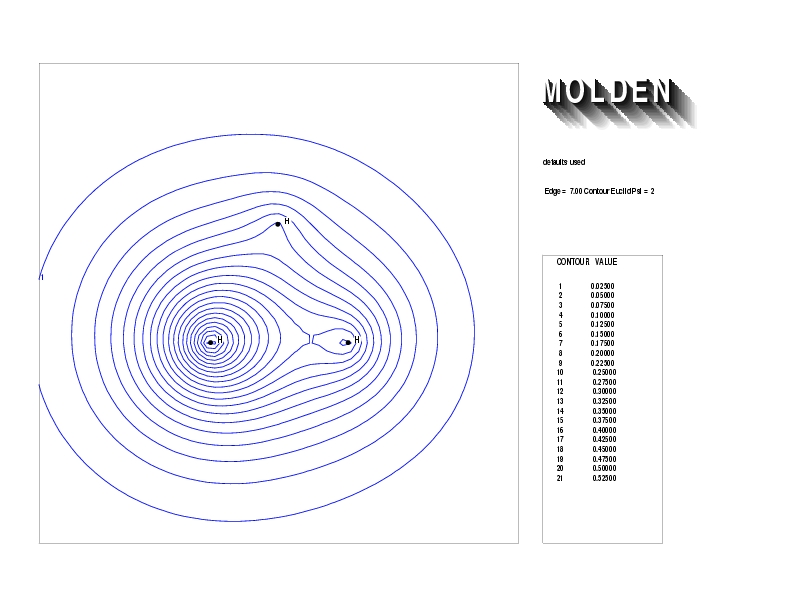
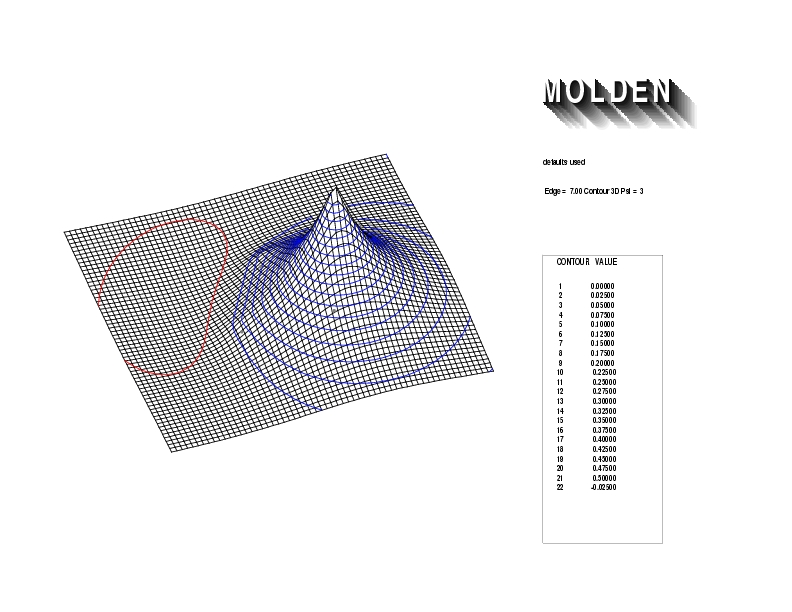
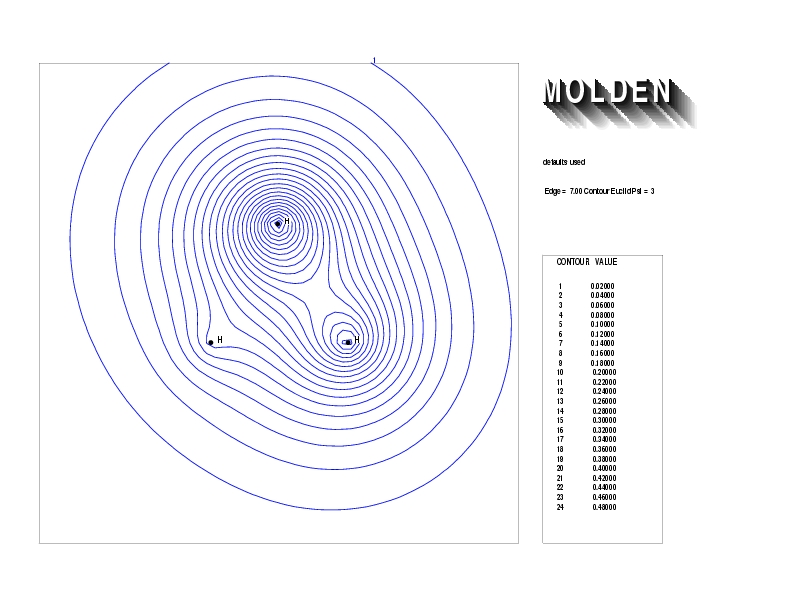
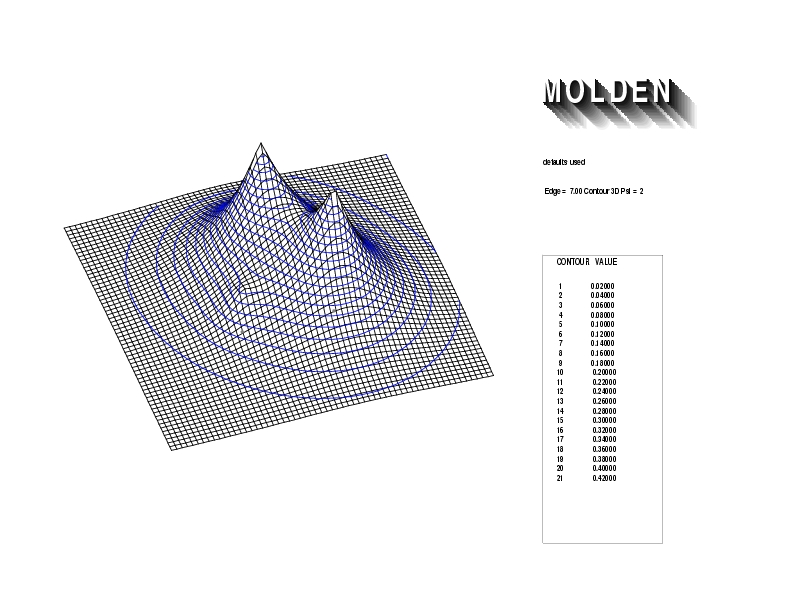
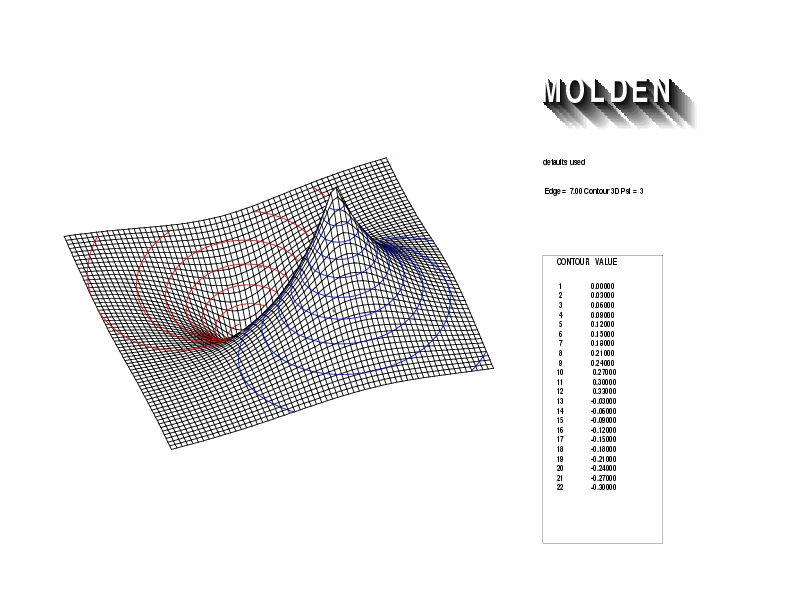
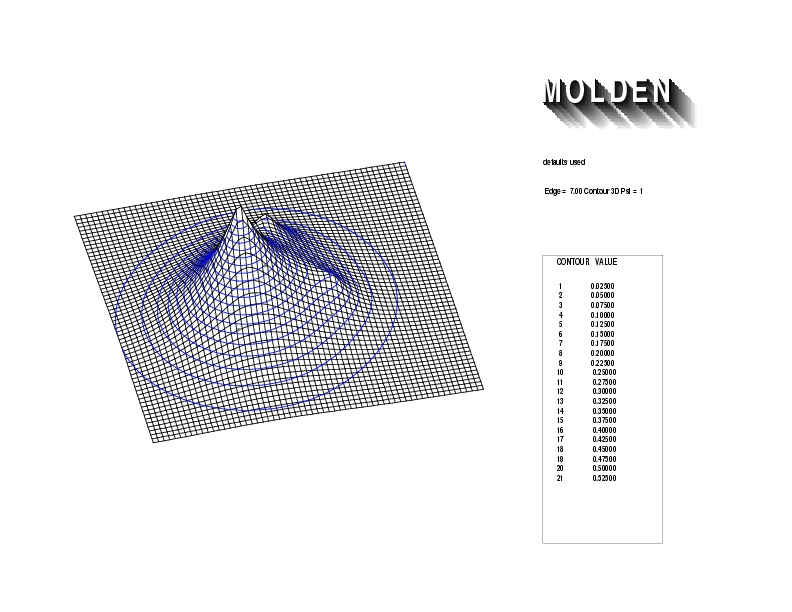
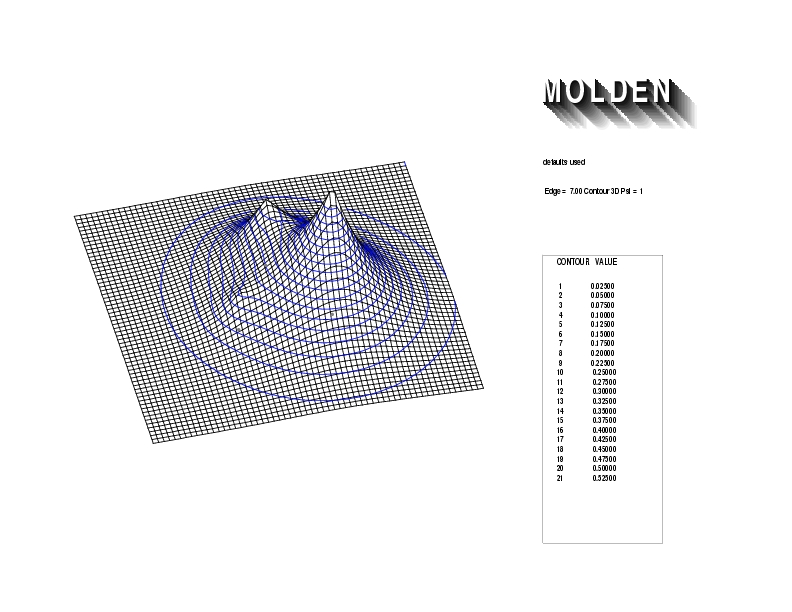
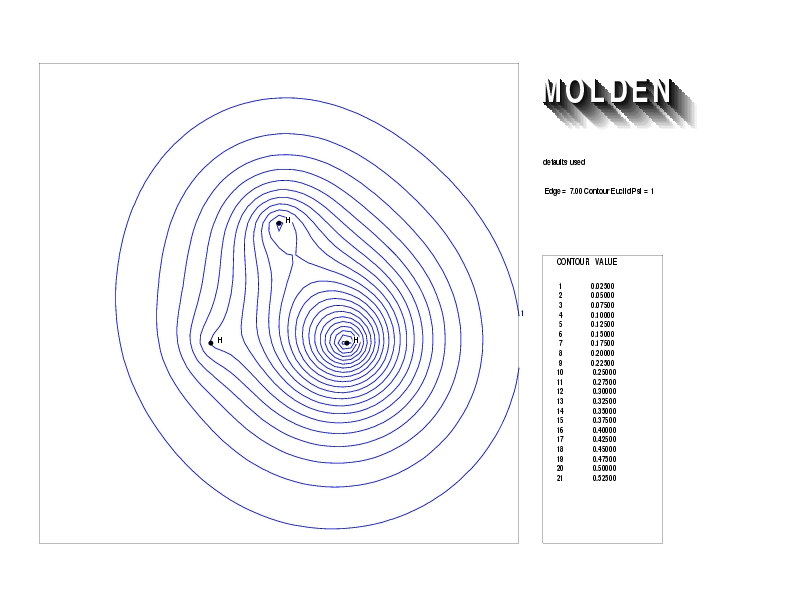
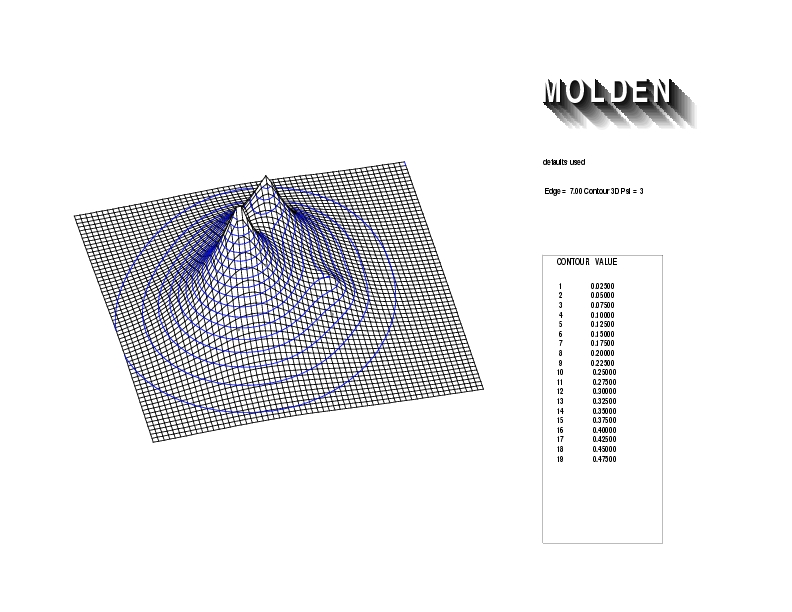
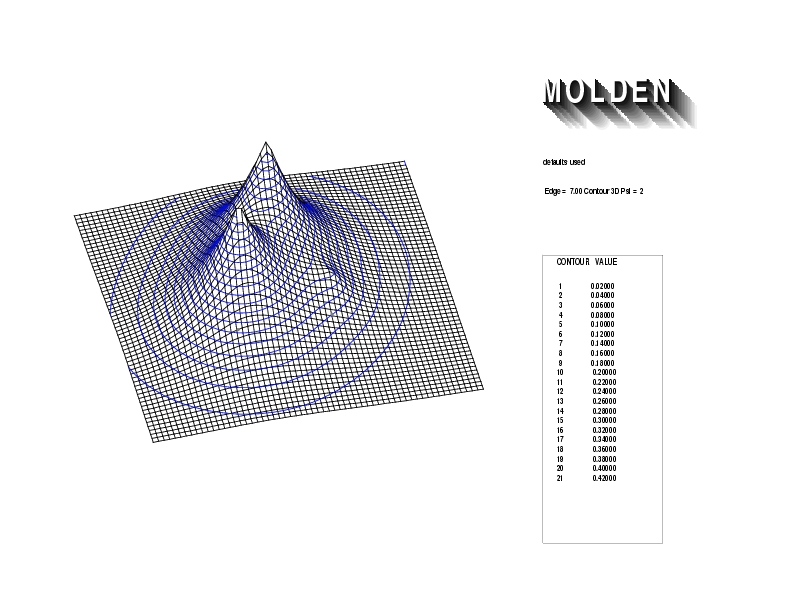
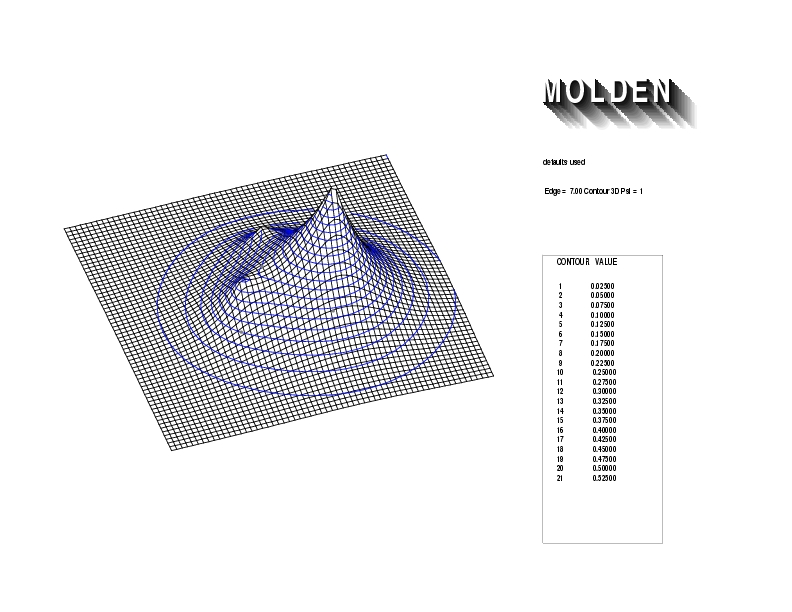
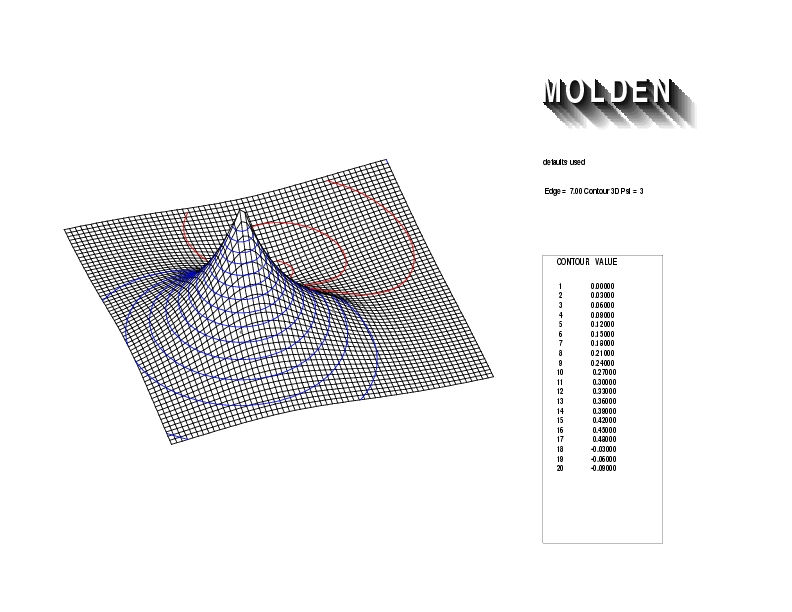
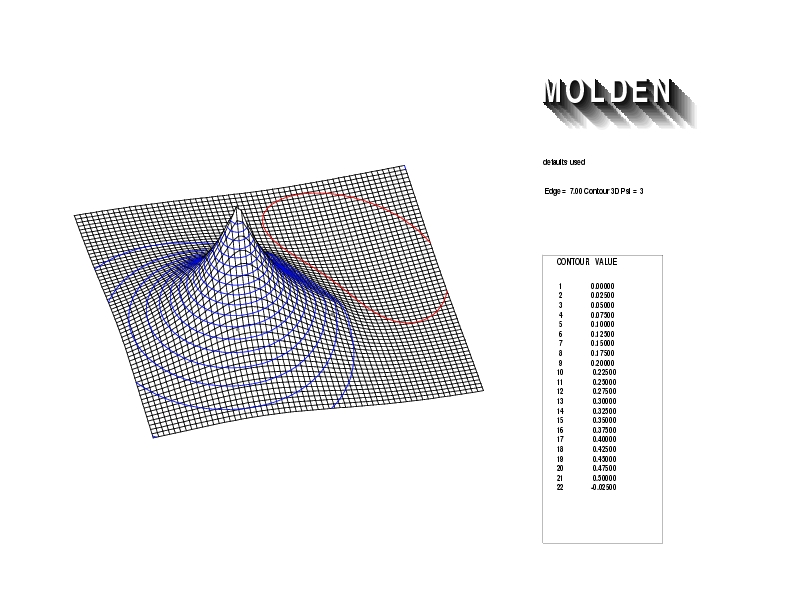
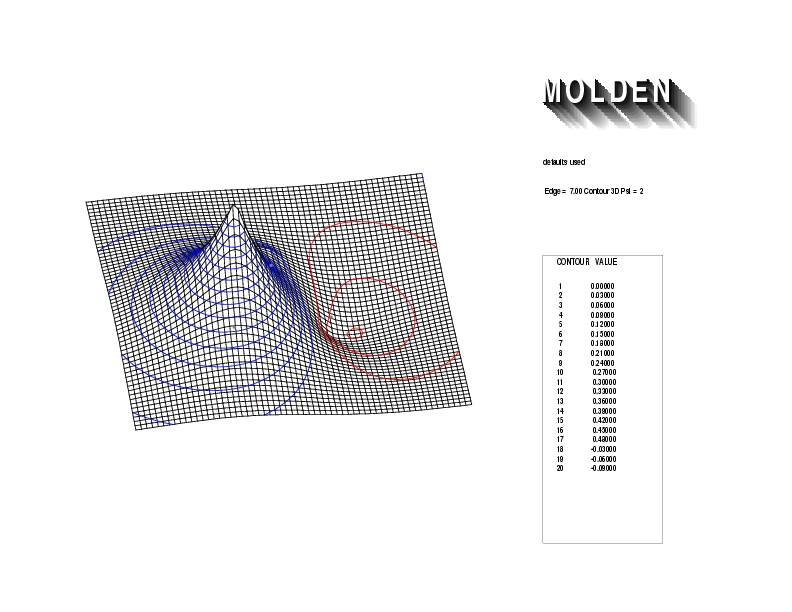
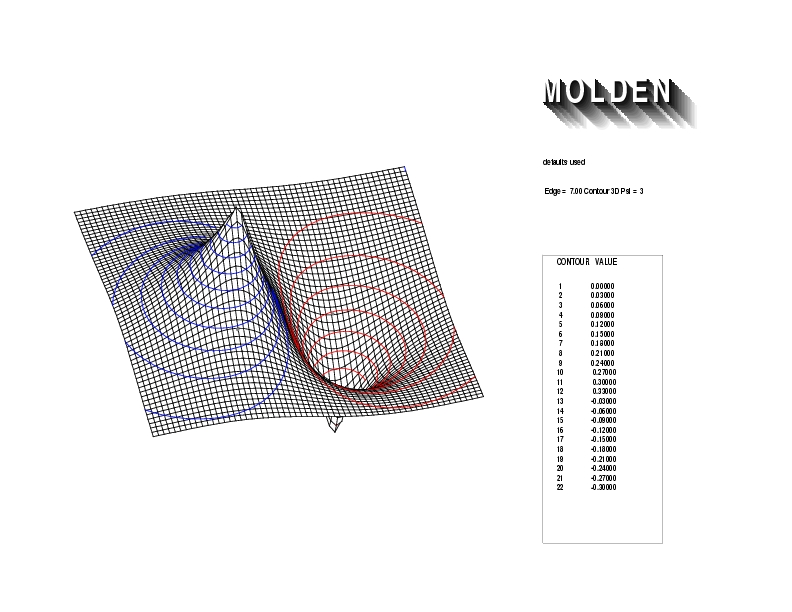
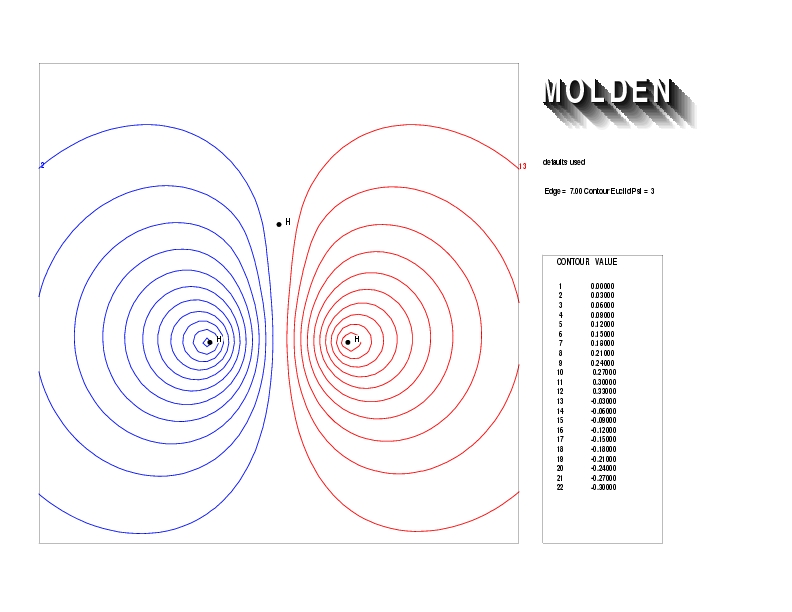
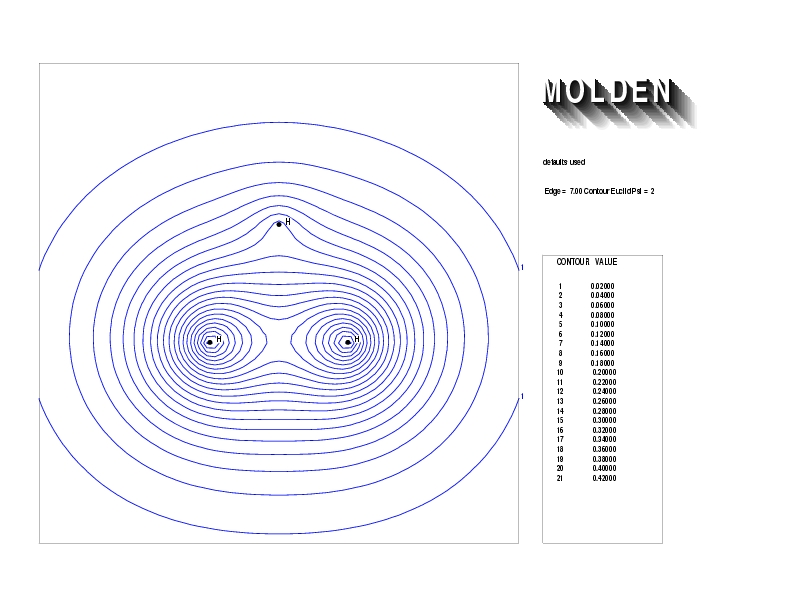
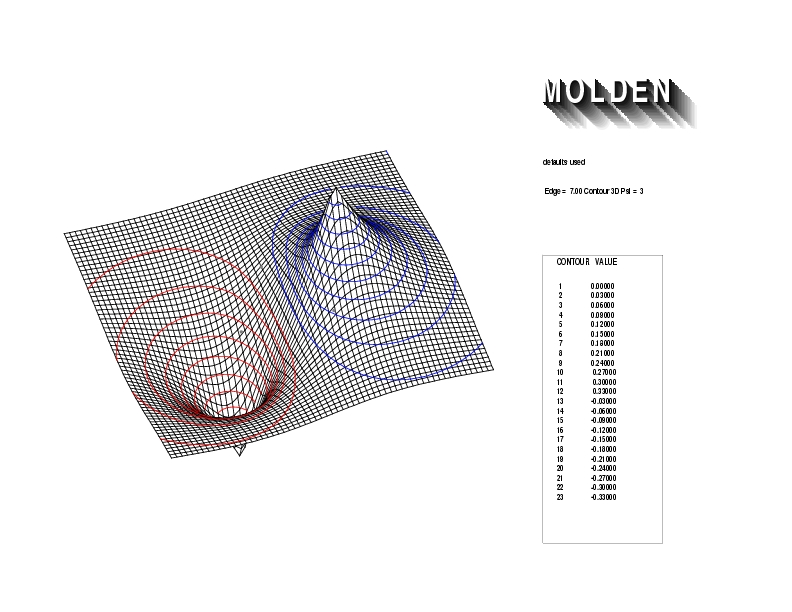
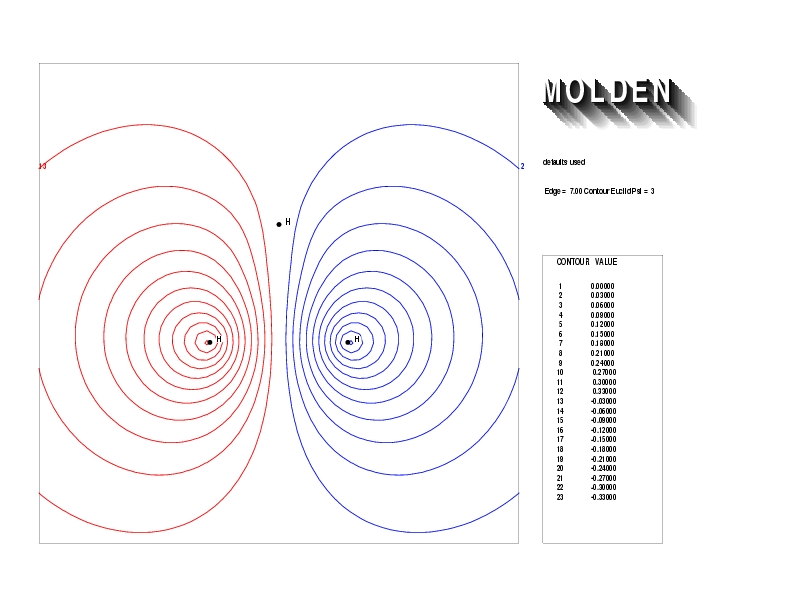
Geometry01: (4.06,3.97,3.97), Spin function=0.2539*Kotani1+0.9672*Kotani2, Energy=-1.537786, Symmetry Group: C2v, Irrep: B2, Orbital 1.
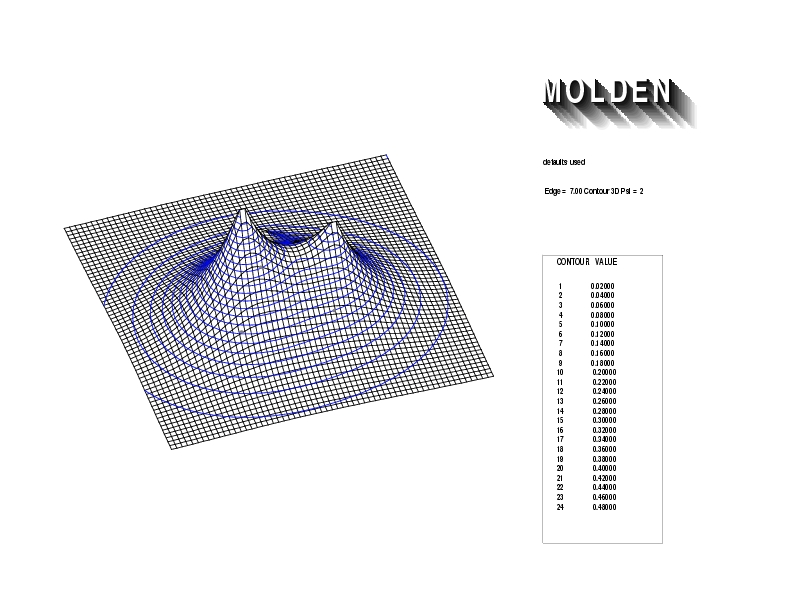
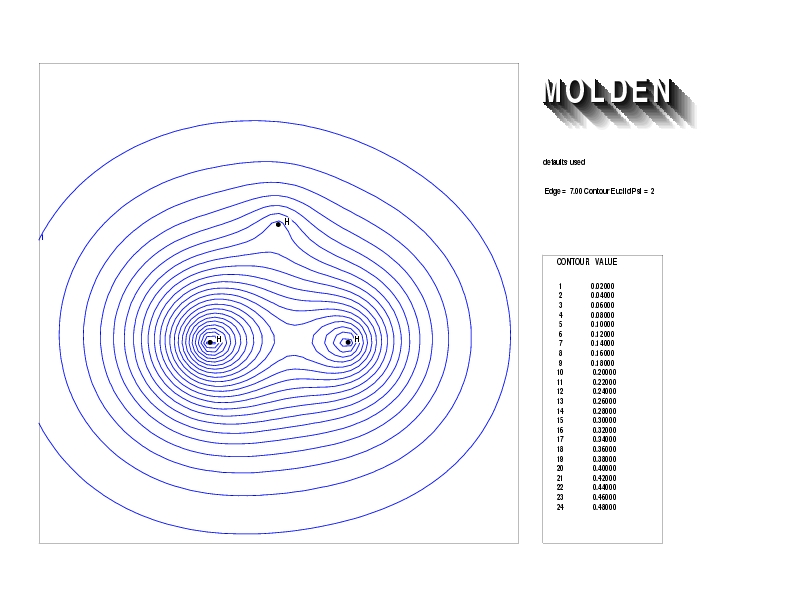
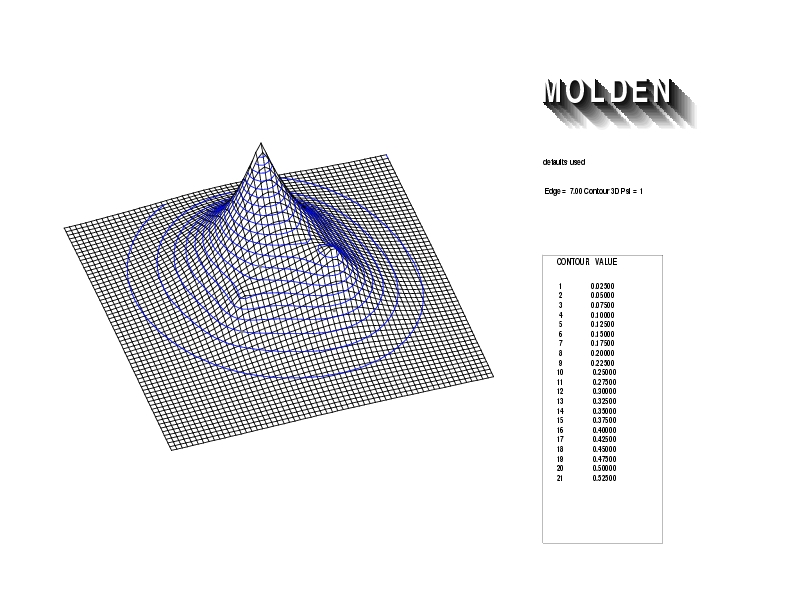
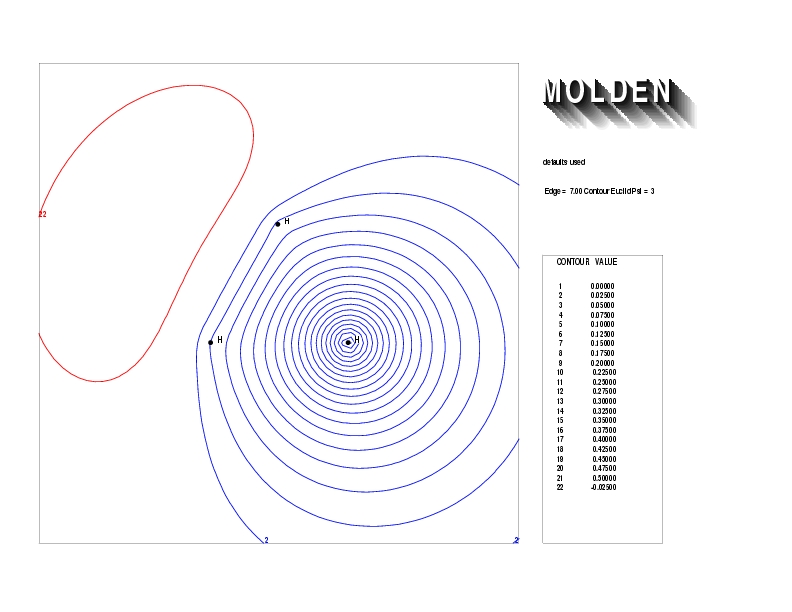
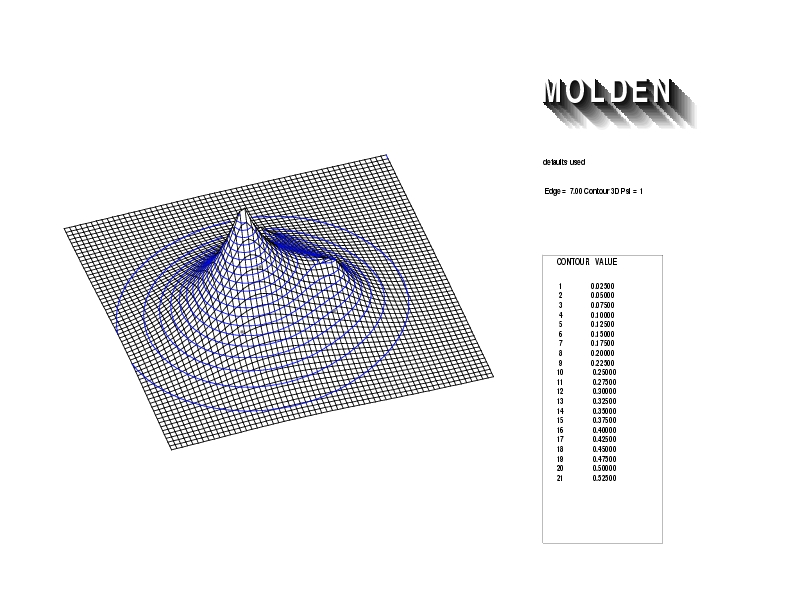
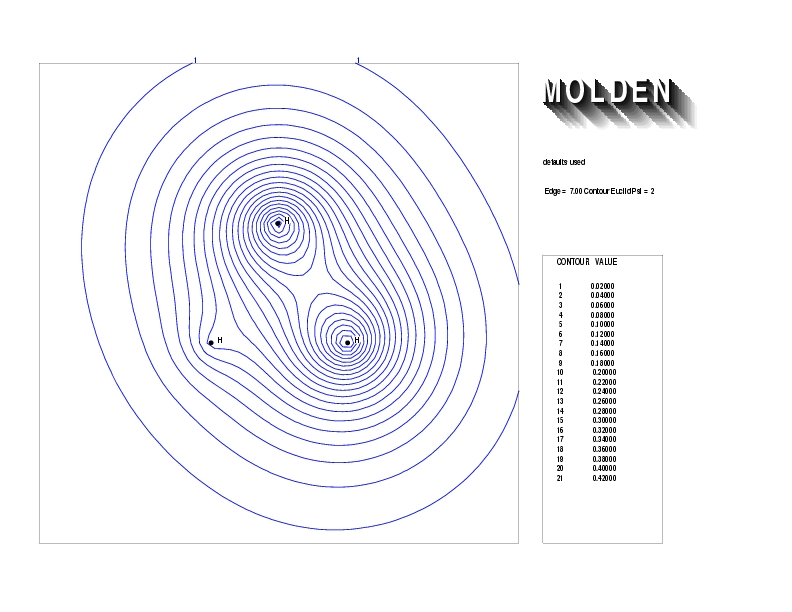
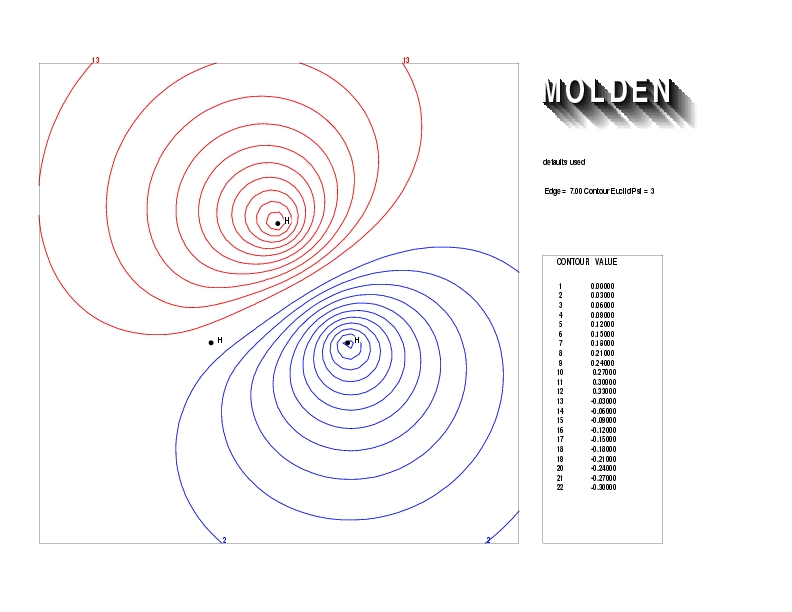
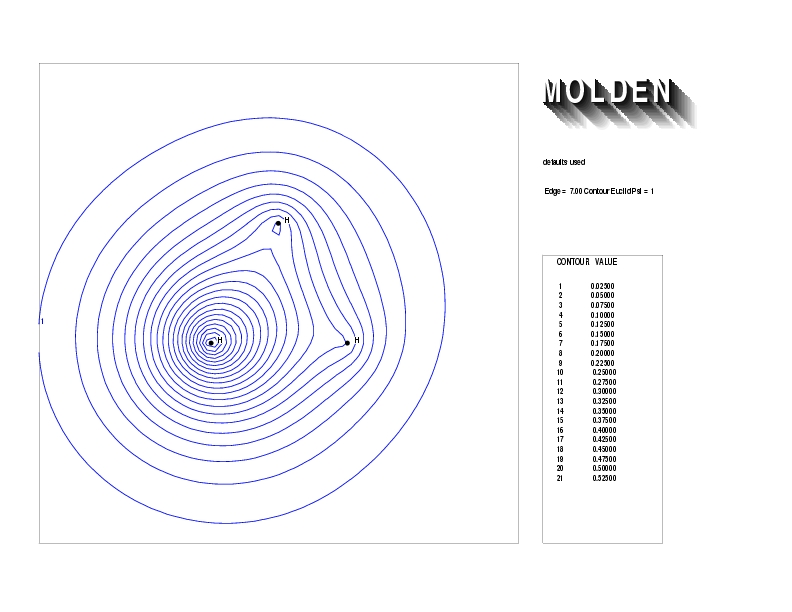
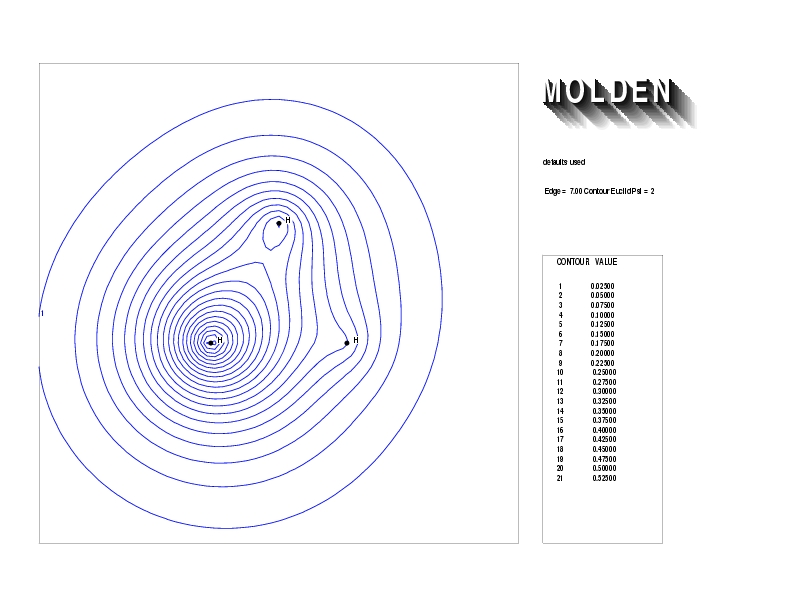
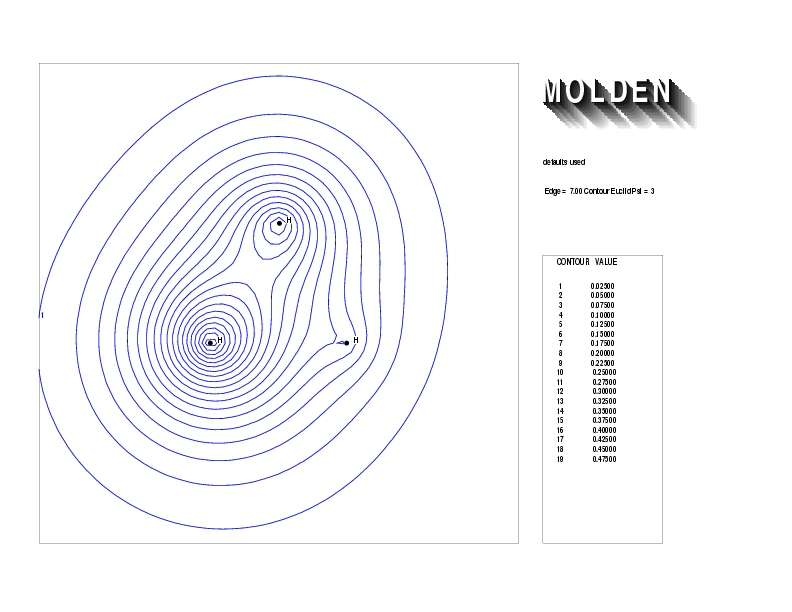
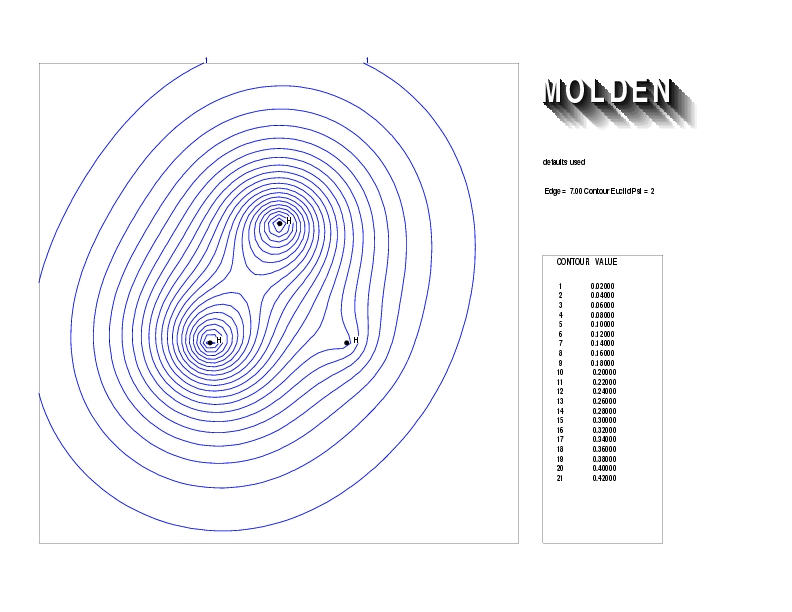
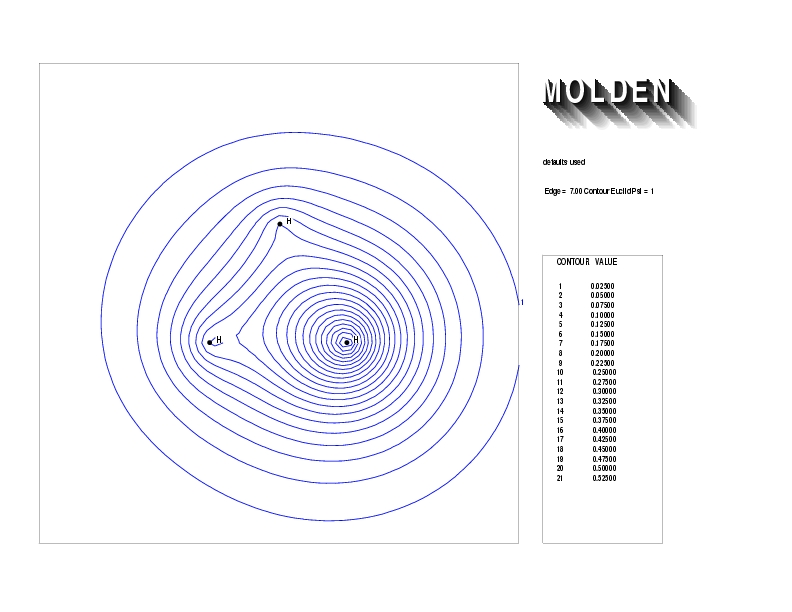
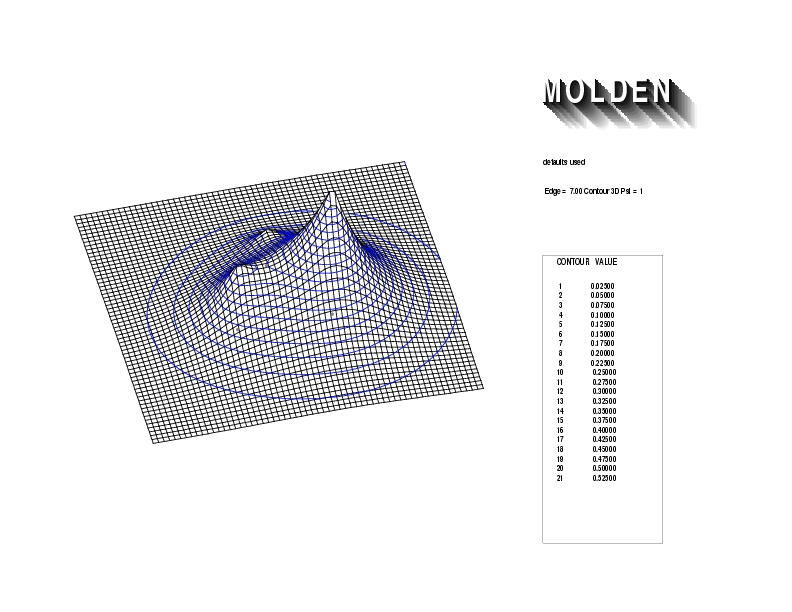
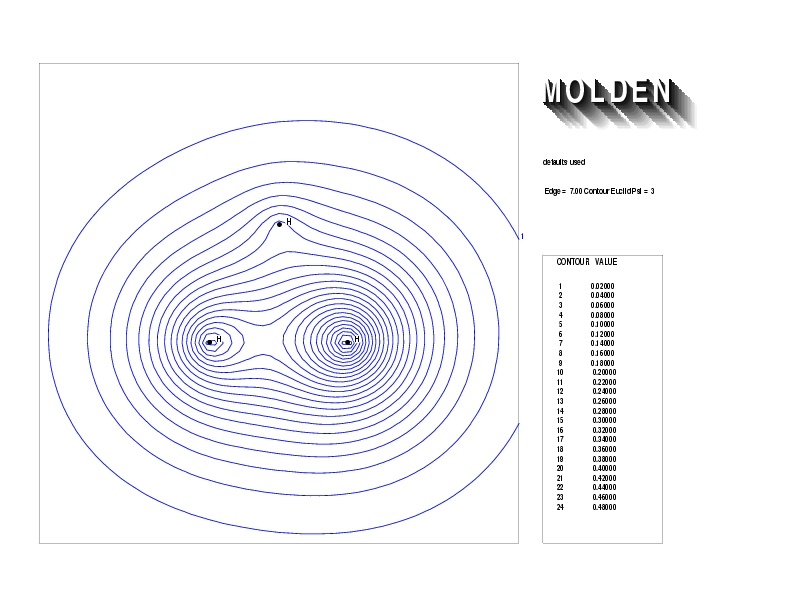
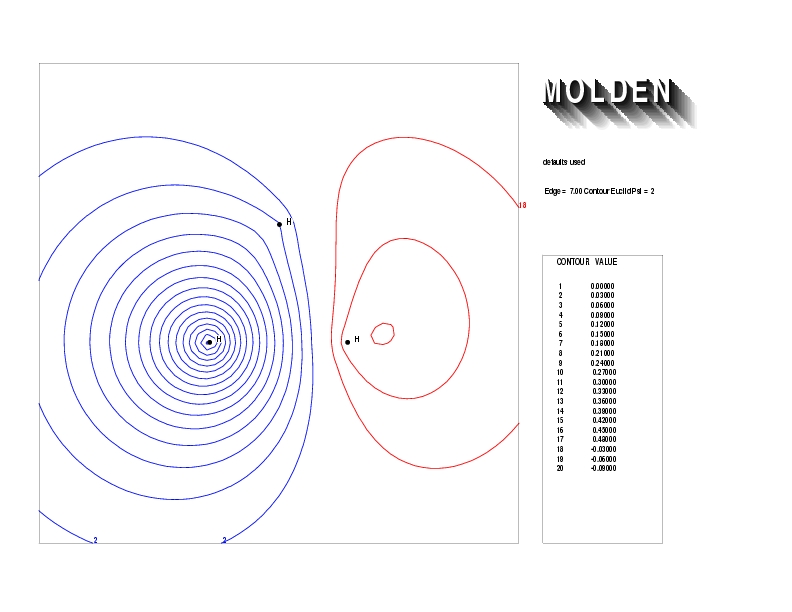
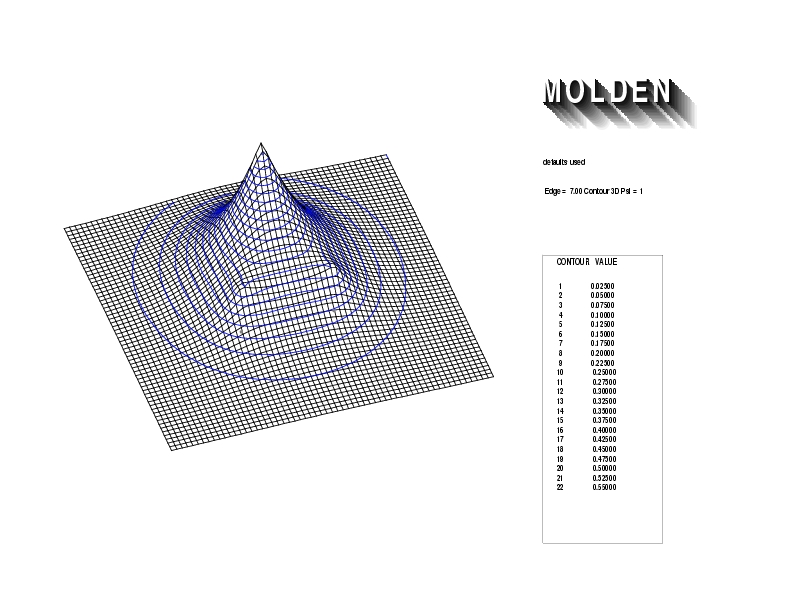
Geometry01: (4.06,3.97,3.97), Spin function=0.2539*Kotani1+0.9672*Kotani2, Energy=-1.537786, Symmetry Group: C2v, Irrep: B2, Orbital 2.
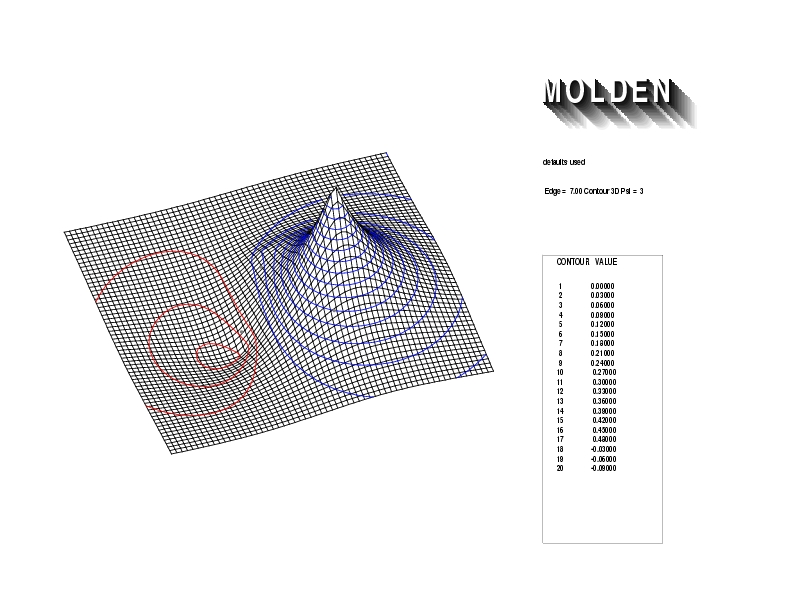
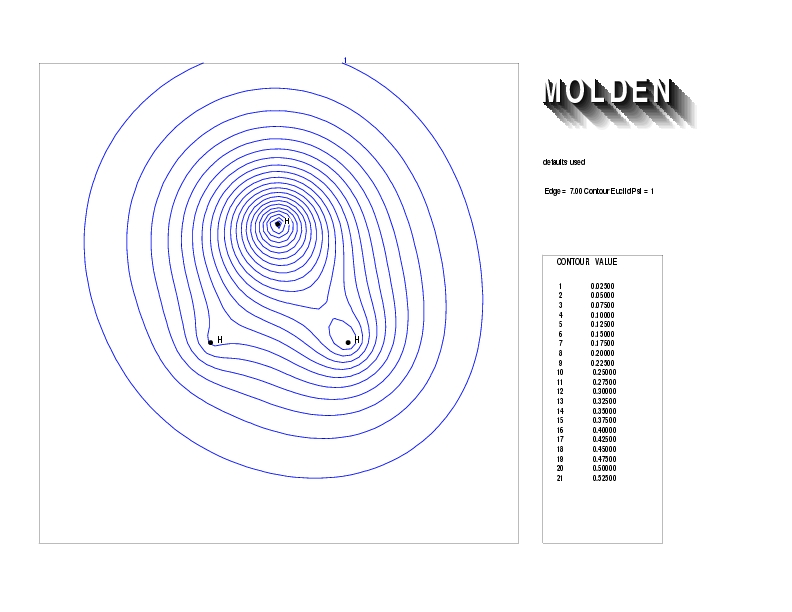
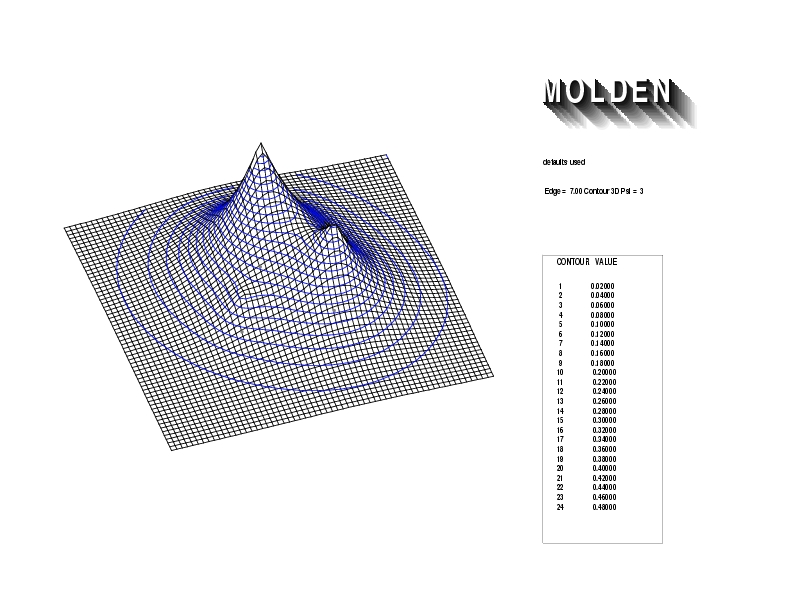
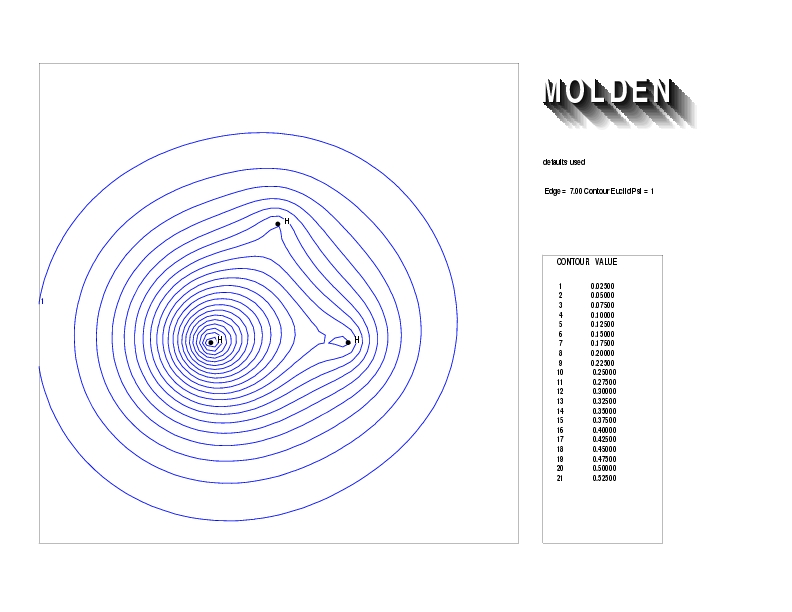
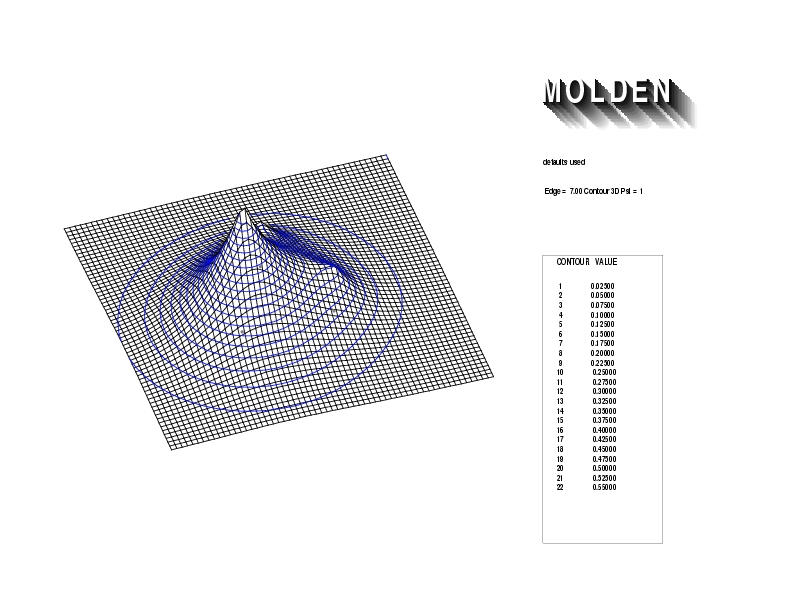
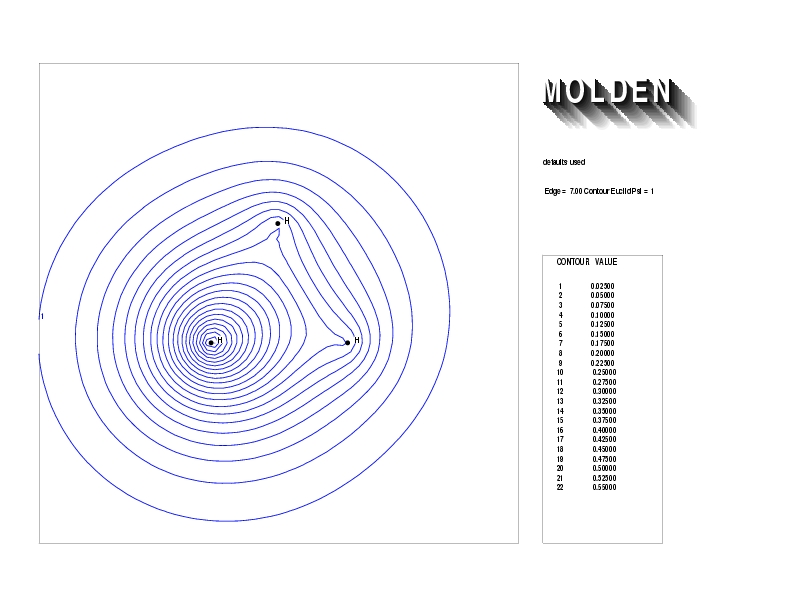
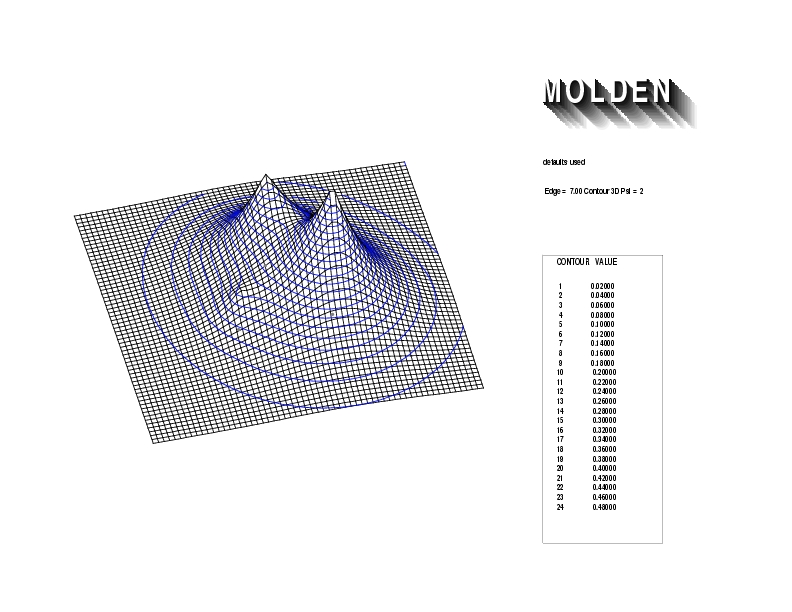
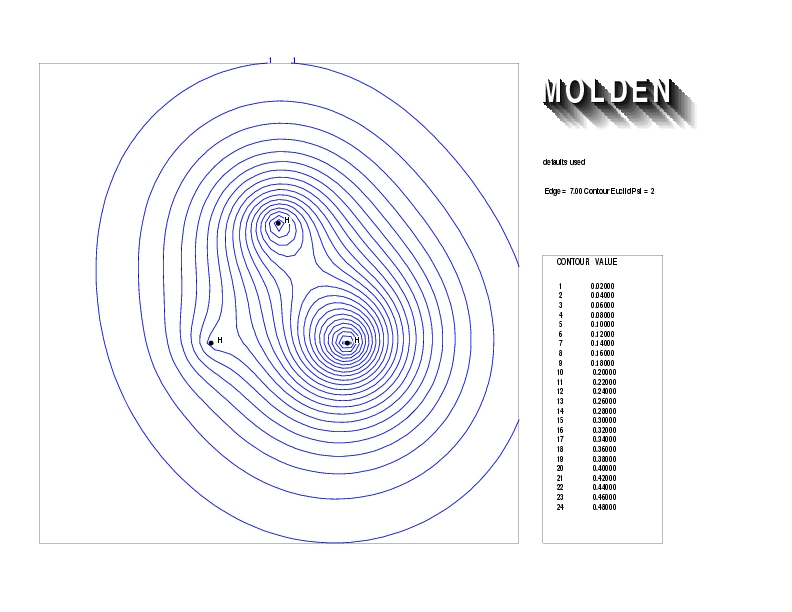
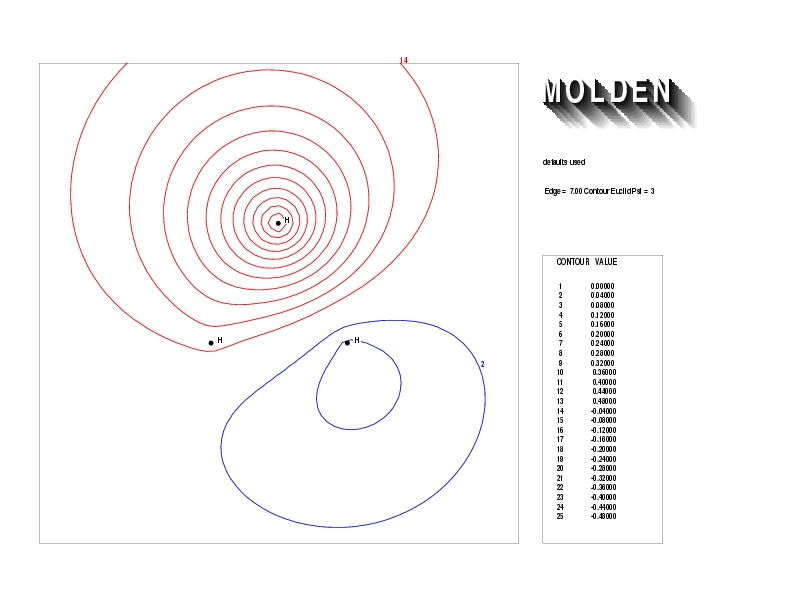
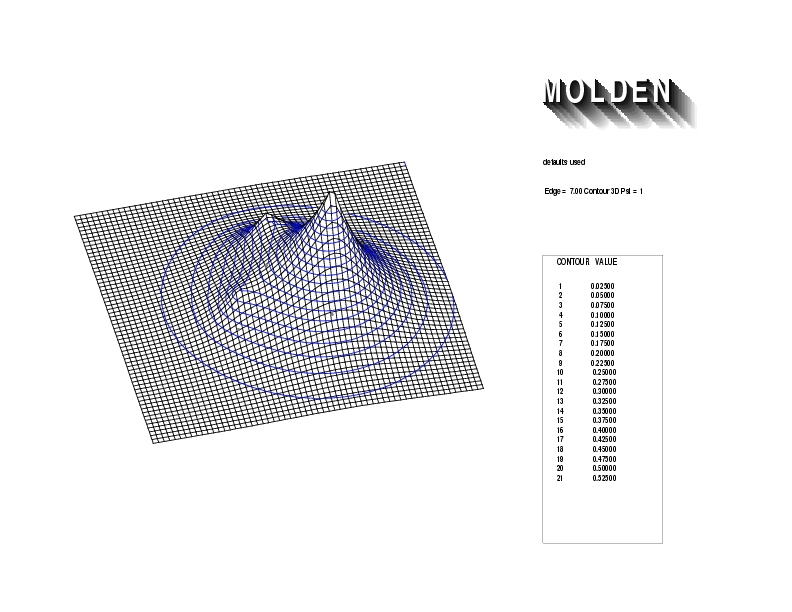
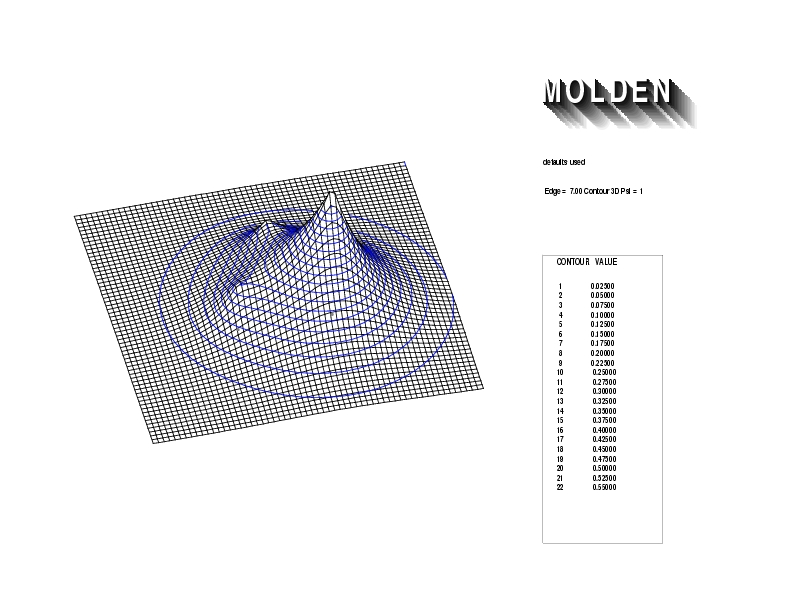
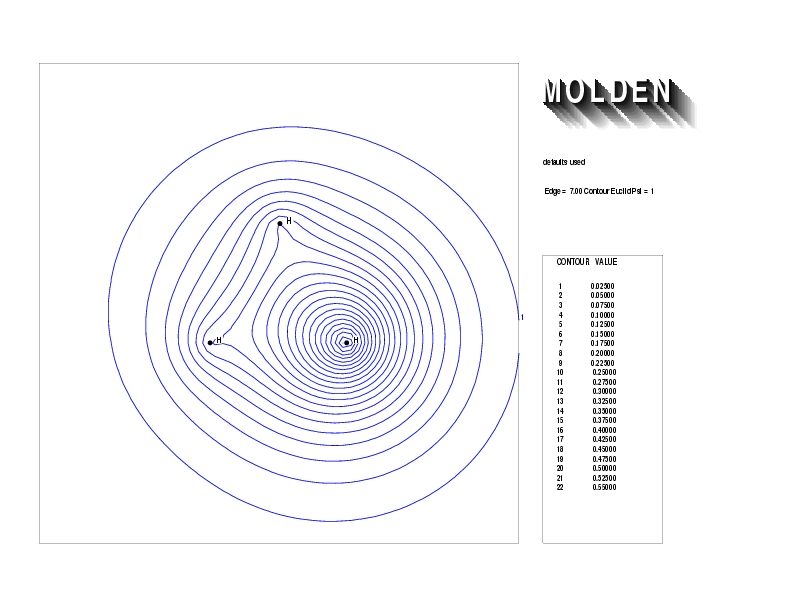
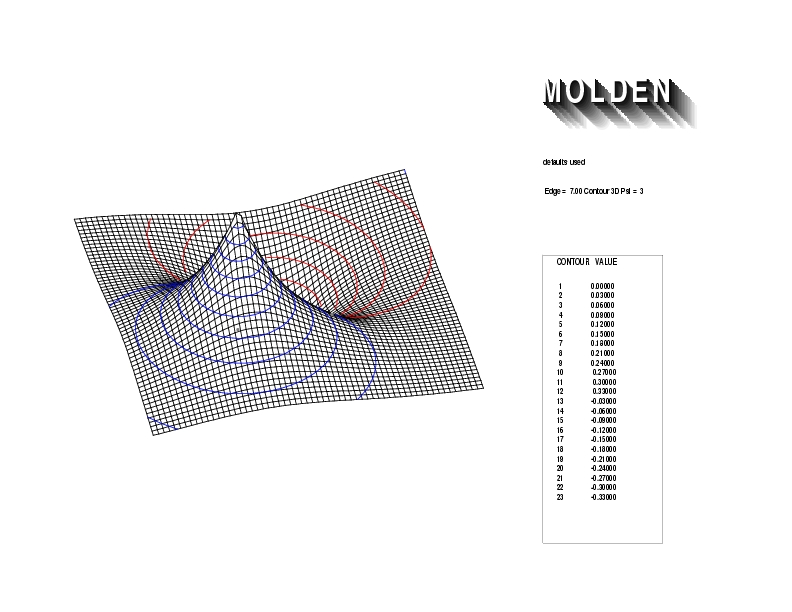
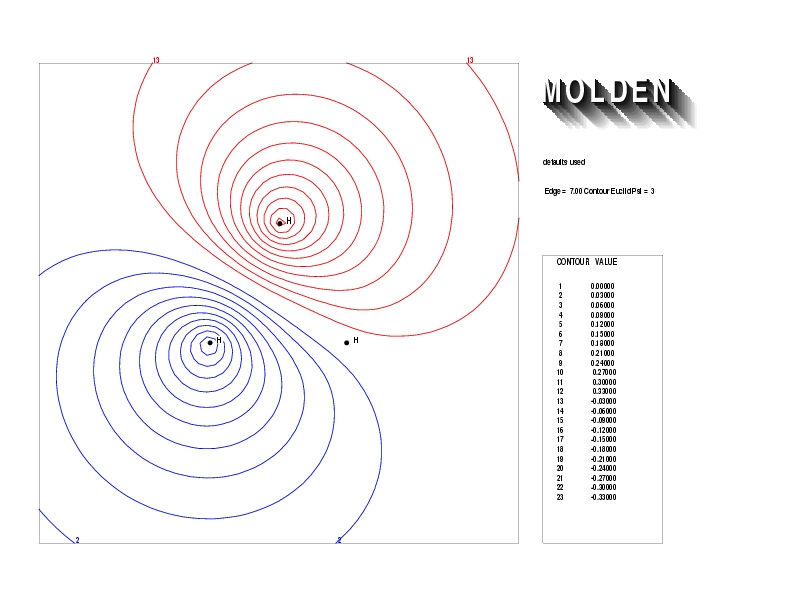
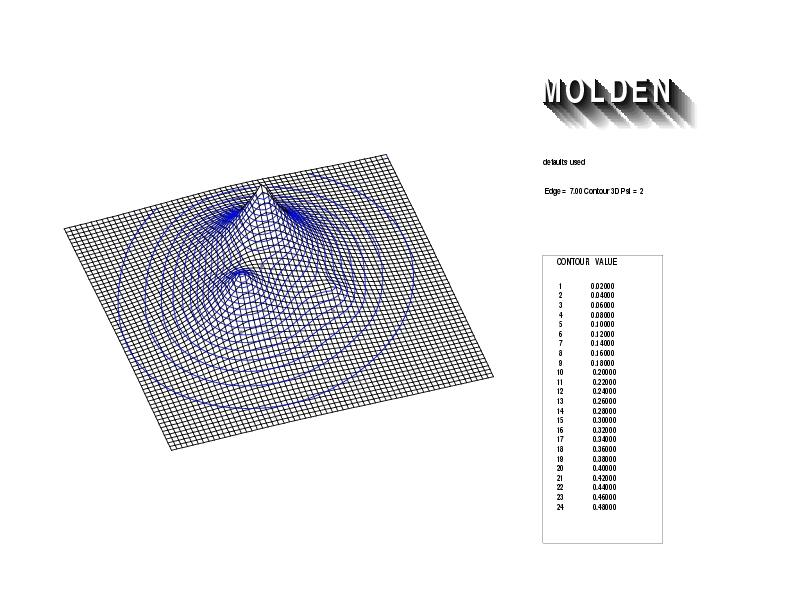
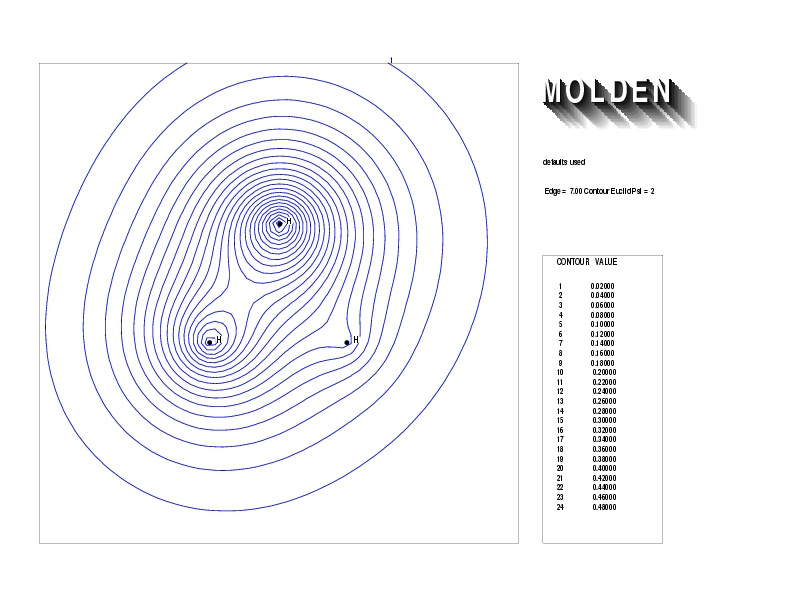
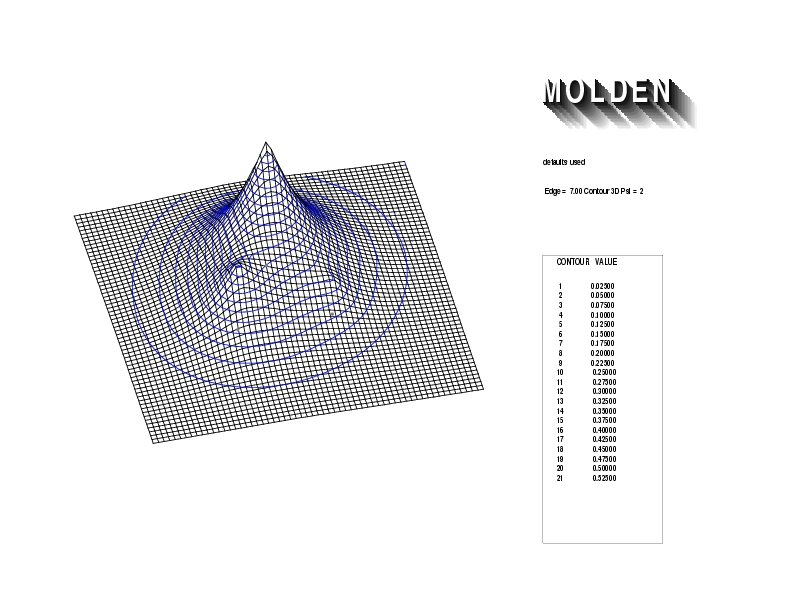
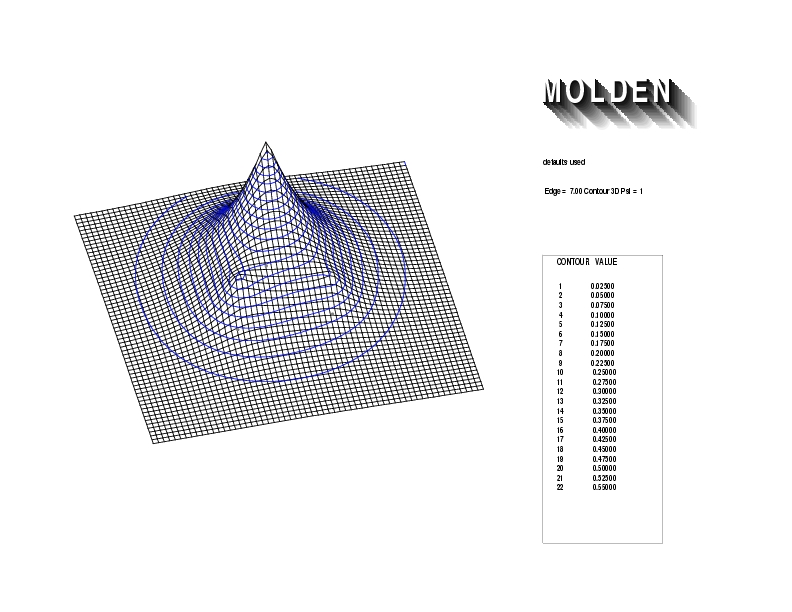
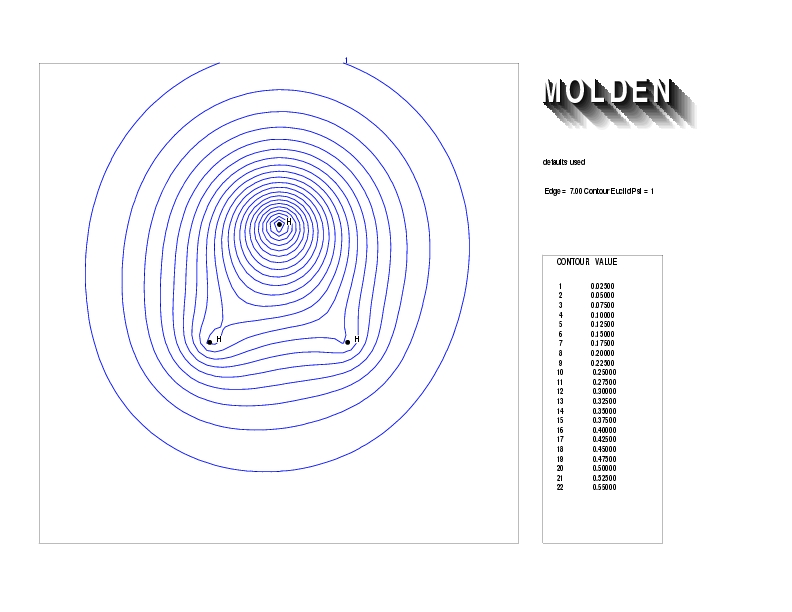
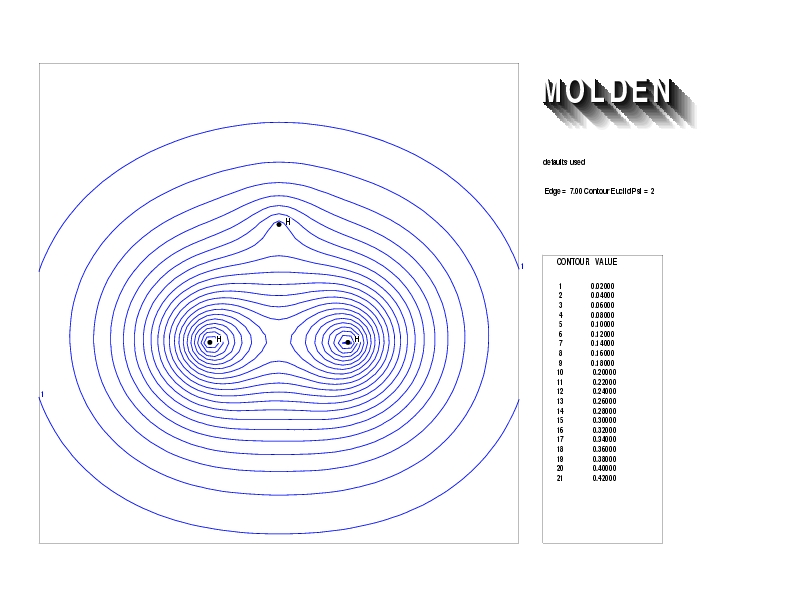
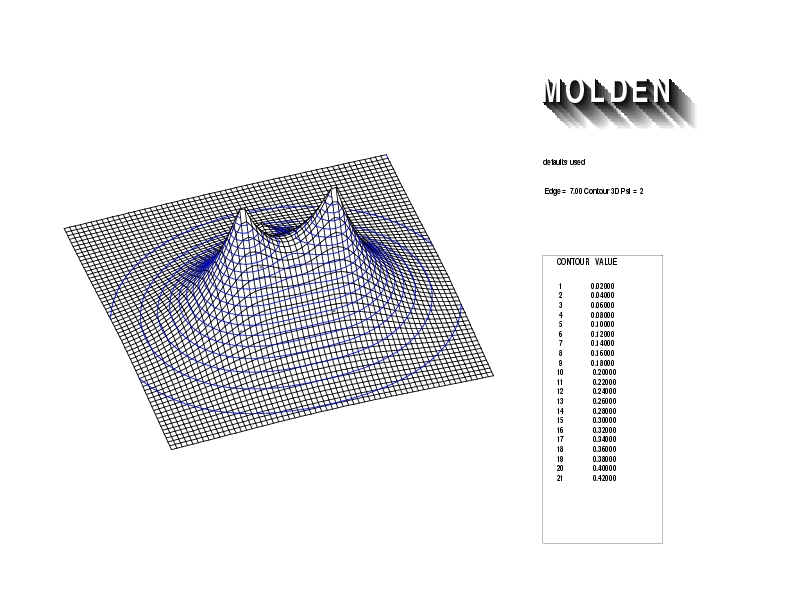
Geometry01: (4.06,3.97,3.97), Spin function=0.2539*Kotani1+0.9672*Kotani2, Energy=-1.537786, Symmetry Group: C2v, Irrep: B2, Orbital 3.
 |
 |
 |
 |
 |
 |